Introduction
Sickle cell disease is an inherited haemoglobinopathy caused by a mutation in the gene encoding the haemoglobin subunit β, encoded by the beta globin gene. It is inherited in an autosomal recessive manner, with HbAS individuals being carriers and HbSS individuals being affected by the disease.
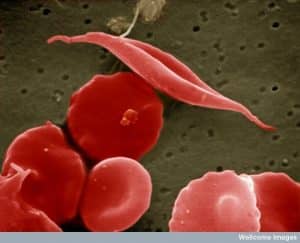
Affected haemoglobin polymerises and forms crystals when deoxygenated. This causes sickling of red cells which can lead to the characteristic vaso-occlusive crises in sickle cell disease sufferers.
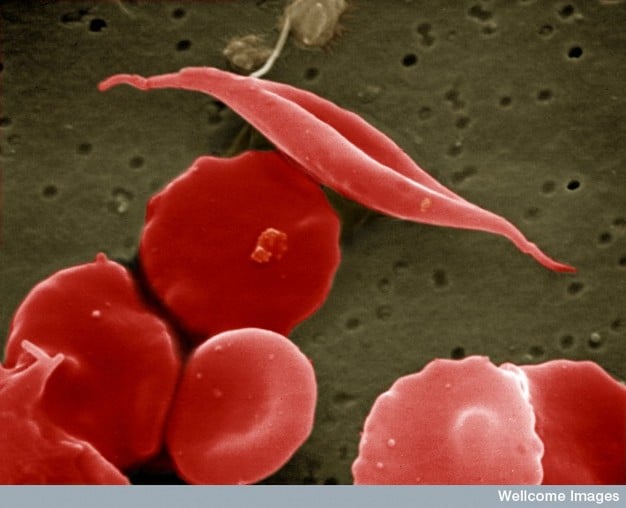
Fig. 1 EM image of sickled red cell
Epidemiology
According to the NHS sickle cell and thalassaemia (SCT) screening programme, the incidence of sickle cell disease is 1 in 2,449 in the UK with a carrier risk of 1 in 89 (1). There is also a marked variation by region with incidences as high as 1 in 861 (London) and as low as 1 in 22,849 or 1 in 10,324 (Northern Ireland and Scotland, respectively).
Globally, the distribution of sickle cell disease also varies. The ‘malaria hypothesis’ that the HbS allele confers a survival advantage against Plasmodium falciparum malaria infection in heterozygotes, suggests the frequency of the HbS allele would follow the distribution of malaria. This has shown to be more consistent in Africa than Asia or the Americas (2). However, with increasing migration, distribution of the HbS allele is now highly heterogenous in non-malaria regions worldwide and data is not reliable enough to give accurate prevalence rates (3).
The burden of sickle cell disease and other haemoglobin disorders is predicted to increase due to higher survival rates amongst infants and increasing prevalence of HbS allele in high-income countries. Thus, more people will survive to adulthood, developing the complications associated with sickle cell disease.
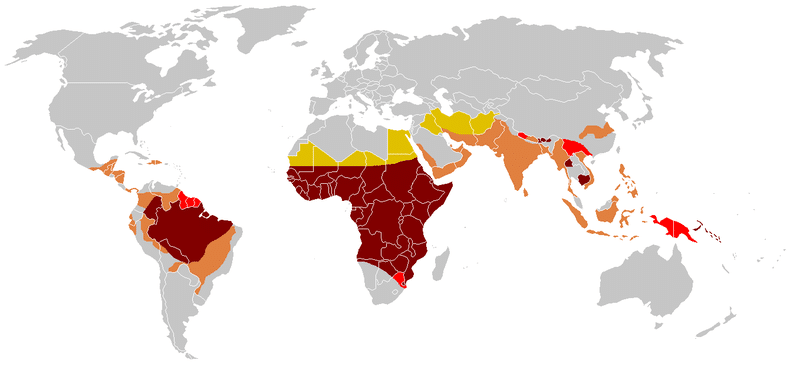
Fig. 2 Global distribution of Sickle Cell Disease
Pathophysiology
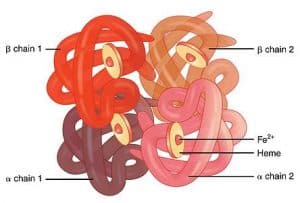
Haemoglobin is made up of 4 globin chains surrounding 4 haem molecules, as follows:
Table 1: Components and prevalence of normal variants of haemoglobin in unaffected, healthy individuals
| Haemoglobin type | Globin chains | % |
| HbA | α2β2 (alpha, beta) | 96.0 – 98.0 |
| HbA2 | α2ω2 (alpha, omega) | 1.5 – 4.0 |
| HbF | α2γ2 (alpha, gamma) | 0.1 – 2.0 |
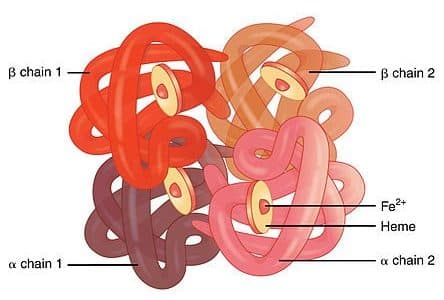
Fig. 3 Graphic showing the 4 globin chains of haemoglobin
The HbS allele results from a single nucleic acid substitution from GAG to GTG in the beta globin gene. This causes glutamic acid to be substituted with valine. Individuals with one HbS allele (HbAS) are carriers while those who are homozygous for the HbS allele (HbSS) have sickle cell disease. This is the most common type of sickle cell disease, however it is also possible to have one HbS allele and a different faulty beta globin allele other than HbS (such as β-thalassaemia, written as HbSβ-thalassaemia).
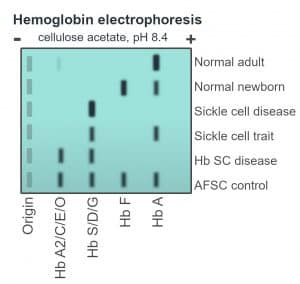
Table 2: Expected results of haemoglobin electrophoresis amongst sickle cell carriers and affected individuals
| Haemoglobin type | HbA | HbF | HbS |
| HbSS | Absent | Variable | 75-95% |
| HbAS | 60% | <2% | 40% |
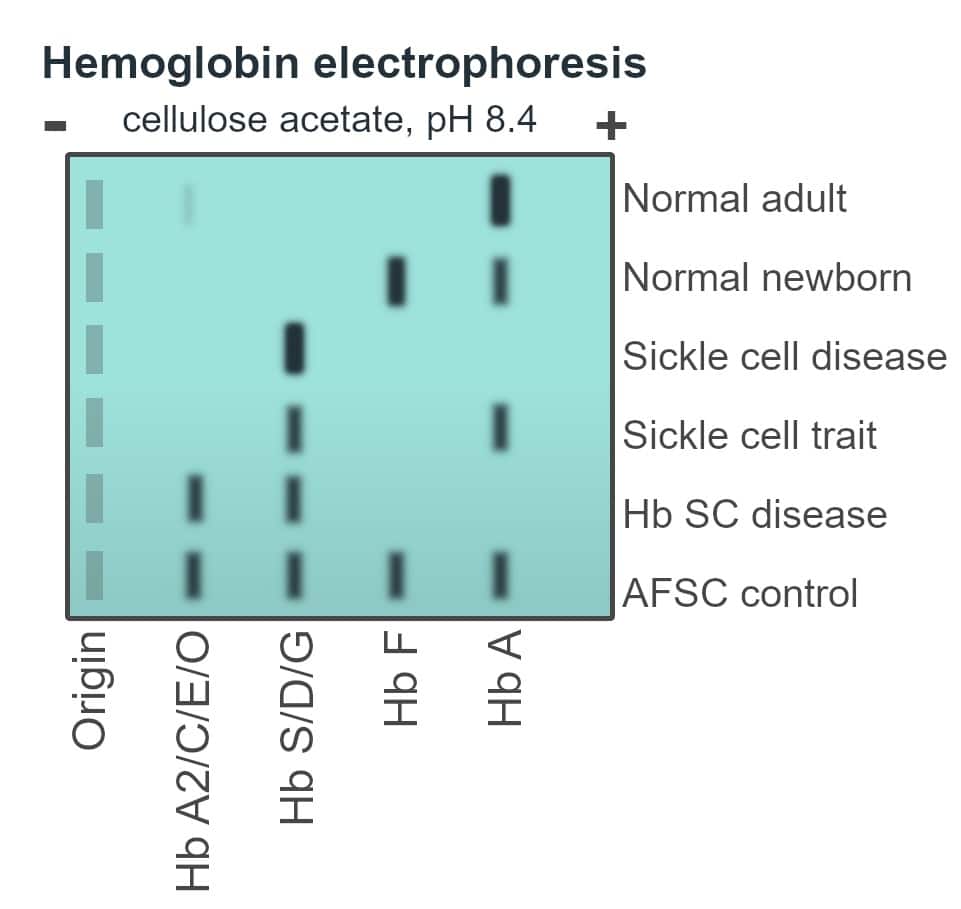
Fig. 4 Schematic of haemoglobin electrophoresis
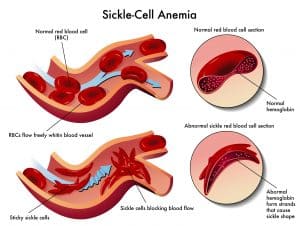
The base pair substitution in the mutated HbS haemoglobin makes it prone to ‘sickling’, where it becomes rigid and distorts into a crescent shape. In the deoxygenated state the HbS tetramers bind to each other and begin to polymerise, growing into long fibres which distort the shape of the red blood cell (4). These sickle cells contribute to vaso-occlusion and chronic haemolysis.
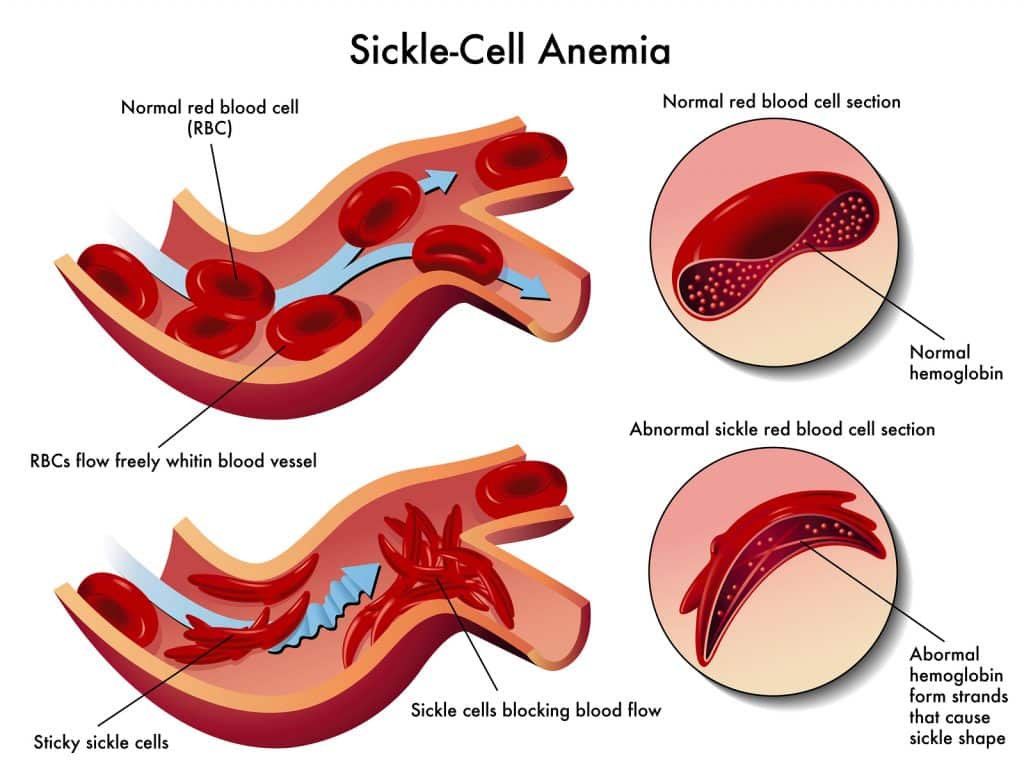
Fig. 5 Graphic demonstrating vast-occlusive crises in sickle cell disease
Risk factors
Ethnicity
In the UK, incidence of sickle cell disease varies markedly by ethnicity as demonstrated below:
Table 3: Incidence of sickle cell disease and carrier status by ethnicity
| Relative incidence | Ethnicity | Affected | Carriers |
| Highest | Black background (eg Black African, Black Caribbean) | 1 in 200 | 1 in 10 |
| Intermediate | Asian | 1 in 6,000 | 1 in 60 |
| Lowest | White | 1 in 90,000 | 1 in 600 |
Clinical features
From history
- Acute pain symptoms from vaso-occlusion – intermittent vaso-occlusive crises are the hallmark of sickle cell disease (5). These episodes are severely painful and may be precipitated by hypoxia, infection, strenuous exercise, dehydration or acidosis (although it is still not completely understood) (6). Vaso-occlusive crises most commonly involve the back, legs, knees, arms, chest and abdomen.
- Dactylitis – exquisitely painful (usually) inflammation of a digit (finger or toe) which may be the first presentation of disease in a child (7).
- Family history – parent diagnosed with sickle cell disease or known carrier.
From examination
- Pallor, lethargy – due to anaemia
- Jaundice – due to haemolysis
- Fever
- Tachycardia, tachypnoea
- Digital redness, swelling and pain – due to inflammation
Differential diagnosis
- Gout – severe, sudden-onset joint pain with redness and swelling
- Acute abdomen – pancreatitis, appendicitis, cholecystitis
When a vaso-occlusive crisis lasts longer than 7 days then it becomes important to consider other causes of bone pain, such as osteomyelitis and avascular necrosis.
Investigations
Laboratory
- Haemoglobin electrophoresis – detects the absence of HbA in sickle cell disease. HbF is higher in newborns and has a protective effect. A patient may first present when HbF decreases at 6 months
- High-performance liquid chromatography
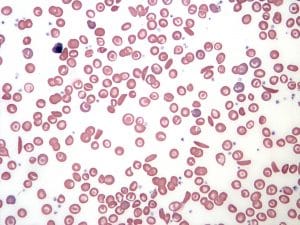
- Blood film – detects the presence of nucleated red blood cells, Howell-Jolly bodies, sickle-shaped red blood cells
- Full blood count and reticulocyte count
- Iron studies
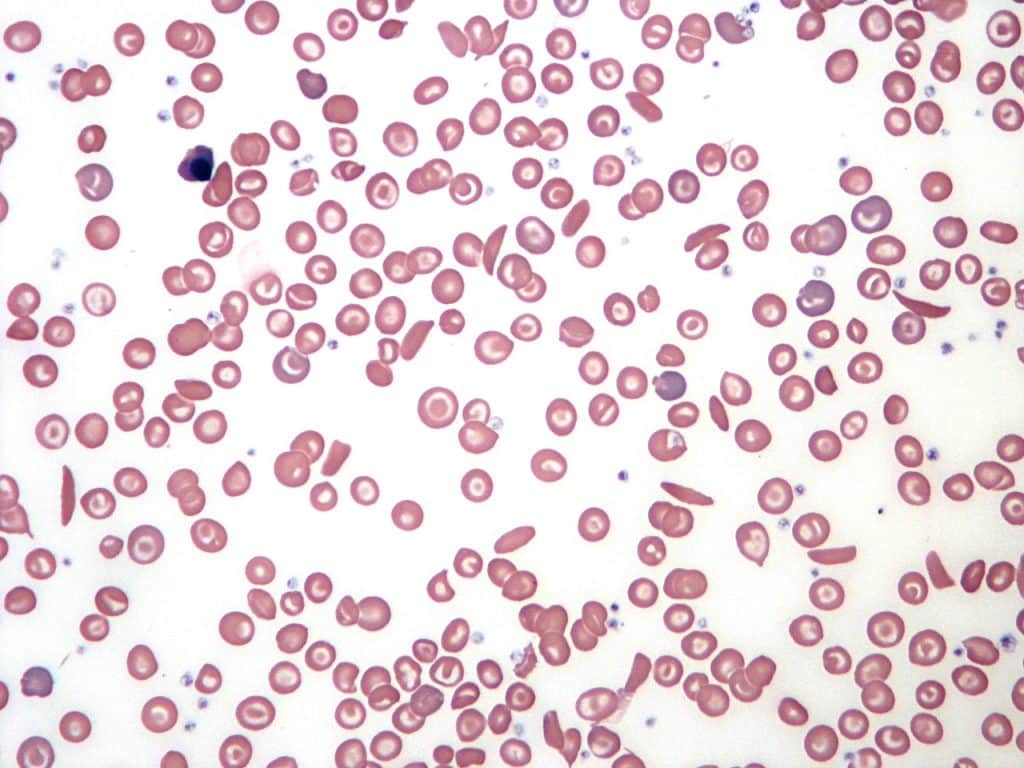
Fig. 6 Blood film in sickle cell anaemia
Imaging
- X-ray of long bones – may be considered to confirm presence of bone infarctions
- Chest X-ray – if chest symptoms as infiltrates may indicate acute chest syndrome
Screening
All pregnant women are offered antenatal screening for sickle cell disease and thalassaemia.
The newborn blood spot test (heel prick test) detects sickle cell disease and thalassaemia. If it is positive, infants are referred for confirmatory testing.
Management
Acute pain crisis
- Analgesia
- Pain should be reassessed regularly
- Paracetamol and NSAIDS may be used for mild pain
- Stronger opioids may be used for moderate to severe pain (8)
- Adequate fluid hydration
- Oxygen
- Antibiotics
- Consider if any evidence of infection
- Follow local protocol
Prophylactic treatment
- Hydroxycarbamide – reduces frequency of painful crises (9)
- Novel and experimental treatments – including L-glutamine, crizanlizumab and Voxelotor (10)
- Regular blood transfusions – targeted to maintain HbS <30% (11)
Education
Patient and caregiver education is important for understanding the disease and recognising potential complications early (12). Education on prevention, for example avoiding excessive alcohol consumption or strenuous exercise, is also an important part of management.
Education for healthcare providers is also essential to allow early diagnosis and management of these patients. This is because a sickle cell crisis can manifest in many ways, such as an acute abdomen, making it an important differential to keep in mind (13).
Complications
Chronic spleen damage by auto-infarction can cause increased susceptibility to infection, particularly pneumococcal and salmonella infections. Therefore, patients should receive regular vaccinations including:
- seasonal influenza vaccines – annual
- pneumococcal vaccines – every 5 years
- hepatitis B vaccine – if receiving regular blood transfusions
- other vaccines, such as meningococcal C or haemophilus influenza type B vaccine – may be advisable if they haven’t had them before
Complications of pulmonary and systemic vasculopathy including:
- pulmonary hypertension
- leg ulcers
- priapism
- chronic kidney disease
- large-artery ischemic stroke (14)
Aplastic crisis can result from infection with parvovirus B19.
Prognosis
According to a study done in 2014 in the US, median life expectancy is 58 years for HbSS and 66 years for HbSC and HbSbeta-thalassaemia (15). Poor prognostic factors include earlier first presentation and high frequency of recurrent vaso-occlusive crises.
Life expectancy is increasing with infection (sepsis, meningitis, pneumonia) or stroke being the commonest causes of death among children and acute chest syndrome among adults (16). However, the proportion of patients dying from infection is decreasing, potentially due to the use of prophylactic penicillin, or better awareness and clinical response to acute presentations (17).
References
- Public Health England. Sickle cell and thalassaemia screening: data report 2018 to 2019. Public Health England. 2021.
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1(8).
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of Sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142–51.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol Mech Dis. 2019;14(1):263–92.
- Manwani D, Frenette PS. Vaso-Occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Vol. 122, Blood. 2013. p. 3892–8.
- Yale SH, Nagib N, Guthrie T. Approach to the vaso-occlusive crisis in adults with sickle cell disease. Am Fam Physician. 2000;61(5):1349–56.
- Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Blood. 1995;86(2):776–83.
- Sickle Cell Society. Sickle Cell Disease in Childhood: Standards and Recommendations for Clinical Care. 2019.
- Lanzkron S, Strouse J, Wilson R, Beach MC, Haywood C, Park H, et al. Systematic Review: Hydroxyurea for the Treatment of Adults with Sickle Cell Disease. Ann Intern Med. 2017;148(12):939–55.
- Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019;381(6):509–19.
- Chonat S, Quinn CT. Current Standards of Care and Long Term Outcomes for Thalassemia and Sickle Cell Disease. Adv Exp Med Biol. 2017;1013:59–87.
- Asnani MR, Quimby KR, Bennett NR, Francis DK. Interventions for patients and caregivers to improve knowledge of sickle cell disease and recognition of its related complications. Cochrane Database Syst Rev. 2014;2014(6).
- Ahmed S, Shahid RK, Russo LA. Unusual causes of abdominal pain: Sickle cell anemia. Best Pract Res Clin Gastroenterol. 2005;19(2 SPEC. ISS.):297–310.
- Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–60.
- Elmariah H, Garrett ME, De Castro LM, Jonassaint JC, Ataga KI, Eckman JR, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530–5.
- Van Der Plas EM, Van Den Tweel XW, Geskus RB, Heijboer H, Biemond BJ, Peters M, et al. Mortality and causes of death in children with sickle cell disease in the Netherlands, before the introduction of neonatal screening. Br J Haematol. 2011;155(1):106–10.
- Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease Charles. Blood. 2004;103(11):4023–7.